 UNG THƯ HỌC
UNG THƯ HỌC
Ung thư trung mô mạch máu (Angiosarcoma)
Đây là nhóm bệnh lý gan ác tính nguồn gốc trung mô phổ biến nhất, chiếm 2% các khối u ác tính tại gan, nhóm tuổi chủ yếu mắc bệnh là 60-70, tỷ lệ nam: nữ là 4:1 [24]. Đây là loại ung thư có tiên lượng xấu, tại thời điểm chẩn đoán hầu hết đã phát hiện di căn, trong đó 60% trường hợp di căn gặp ở phổi và lách, thời gian sống thêm trung bình là 6 tháng [25].
Hình thái tổn thương chia làm 4 nhóm chính: đa nốt, khối u lớn, dạng hỗn hợp (khối lớn kèm đa nốt) và dạng thâm nhiễm. Trong đó, dạng đa nốt và thâm nhiễm chiếm tỷ lệ cao nhất [26].
Thương tổn trên chẩn đoán hình ảnh thường rất đa dạng, phù hợp với tính đa dạng trong đặc điểm giải phẫu bệnh của loại ung thư này [5]. Trên siêu âm, hình thái thể hiện là một khối u dạng máu, thường là vùng tăng âm kèm theo bóng cản phía sau thể hiện tình trạng calci hóa [6].
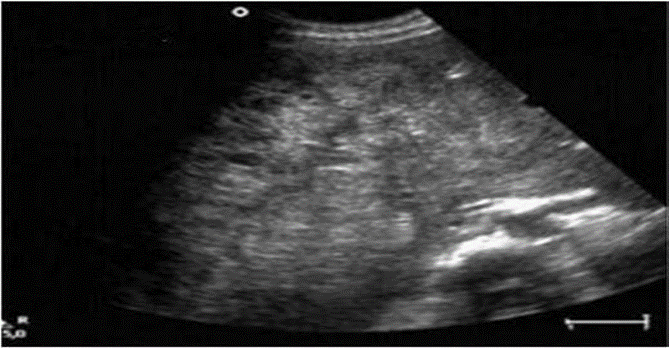
Hình 18: Angiosarcoma. Hình ảnh siêu âm là khối tăng cản âm tương đối, hỗn hợp âm chiếm toàn bộ thùy gan phải.
Trong chụp cắt lớp vi tính không tiêm thuốc, khối u có đặc điểm giảm tỷ trọng, tuy nhiên có thể gặp những vùng tăng tỷ trong do hiện tượng chảy máu trong u. Sau tiêm thuốc, tại pha động mạch (Hình A và B) và pha tĩnh mạch cửa (Hình C), xuất hiện những vùng tăng tỷ trọng dạng mảnh hoặc nhẫn, thể hiện tình trạng tăng sinh mạch trong u, duy trì và tiến triển đến thì muộn (Hình D) [24]. Các nghiên cứu cho thấy ung thư trung mô mạch máu không thể hiện tình trạng tăng ngấm thuốc dạng hướng tâm như loại u máu do dị dạng mạch (cavernous hemangiomas) [24].




Hình 19: Angiosarcoma. Hình ảnh cắt lớp vi tính ổ bụng không tiêm thuốc thể hiện hình ảnh khối giảm tỷ trọng hoặc tỷ trọng hỗn hợp, xen kẽ nốt vôi hóa chiếm toàn bộ thùy gan phải (Hình A). Ở thì động mạch, có tình trạng tăng ngấm thuốc dữ dội phản ánh sự tăng sinh mạch (Hình B), duy trì đến thì tĩnh mạch cửa (Hình C) và thì muộn (Hình D)
Trên cộng hưởng từ, khối u này thể hiện là những vùng tăng tín hiệu trên chuỗi xung T1, không đồng nhất trên chuỗi xung T2 với những điểm chảy máu, vách ngăn và các điểm vôi hóa xen kẽ, thể hiện trong giai đoạn sớm và giai đoạn chính sau tiêm thuốc [25].
Chẩn đoán phân biệt chính bao gồm Ung thư nội mạc mạch máu, ung thư đường mật trong gan và ung thư gan thứ phát dạng tăng sinh mạch [24].
Như vậy tổng kết những yếu tố hướng tới ung thư trung mô mạch máu bao gồm:
- Nam giới từ 60-70 tuổi
- Tiền sử tiếp xúc với hóa chất như: Thorium dioxide, Vynil chloride, arsenic và đồng chất của Steroids.
- Những khối u không đồng nhất, tăng tỷ trọng với tính chất ngấm thuốc bền vững.
Ung thư nội mạc mạch máu (Epitheliod Hemangioendothelioma)
Đây là loại ung thư có nguồn gốc mạch máu, gặp chủ yếu ở người lớn, đặc biệt là phụ nữ, do đó Estrogen có thể là yếu tố nguy cơ liên quan. Đây là dạng ung thư có nhiều độ biệt hóa, từ mức độ ác tính thấp đến cao. Tính trung bình, tỷ lệ sống thêm 5 năm là xấp xỉ 50% [27].
Dấu hiệu lâm sàng thường không đặc hiệu, bao gồm đau hạ sườn phải, sụt cân, một số trường hợp gây ra viêm gan tối cấp hoặc hội chứng Budd-Chiari [28]. Những vị trí di căn thường gặp nhất bao gồm hạch ổ bụng, phúc mạc, phổi và xương [29].
Hình thái trên chẩn đoán hình ảnh gồm 2 dạng: Dạng nốt và dạng lan tỏa. Dạng đa vi nốt hay gặp trong giai đoạn đầu, với những đa nốt nhỏ dưới cỏ khó phát hiện trên lâm sàng. Giai đoạn sau, các nốt phát triển và hợp nhất, tạo thành các khối u lớn và có tính chất xâm nhập [30].
Trên cắt lớp vi tính có tiêm thuốc cản quang, dạng u này thường có tình trạng ngấm thuốc ngoại vi và trung tâm, cùng với tình trạng co kéo lớp vỏ và canxi trung tâm [16]. Alomari và cộng sự đã mô tả dấu hiệu kẹo mút (‘Lollipop sign’) quan sát được trên cắt lớp vi tính và cộng hưởng từ, gây ra bởi sự chấm dứt đột ngột của tĩnh mạch cửa, động mạch gan trước một khối u lớn ngoại vi. Đây có thể coi là triệu chứng đặc hiệu của dạng tổn thương này [31].




Hình 20: Epithelioid hemangioendothelioma. Phim chụp cắt lớp vi tính không tiêm thuốc cho thấy hình ảnh các nốt giảm âm ngoại vi gan xem kẽ các nốt vôi hóa (Hình A), xảy ra trong 25% trường hợp. Cắt lớp vi tính ngực trên cùng bệnh nhân cho thấy nốt thứ phát viền nhu mô phổi (Hình B). Với cắt lớp vi tính có tiêm thuốc thì tĩnh mạch cửa (Hình C, D), các nốt tại gan phân biệt rõ với vùng nhu mô gan thường, cùng với đặc điểm co kéo bao gan (mũi tên hình C), bên cạnh nốt di căn phổi vôi hóa (mũi tên hình D)
Di căn phổi thường có tình trạng canxi hóa, trong khi di căn xương gây ra tình trạng hủy xương, xâm nhập và ít thay đổi.




Hình 21: Epithelioid hemangioendothelioma. CT ổ bụng tiêm thuốc thì tĩnh mạch cửa cho thấy những nốt giảm sinh mạch ngoại vi (Hình A). MRI cho thấy hình ảnh các nốt nhu mô gan giảm tín hiệu trên chuỗi xung T1 (Hình B) và tăng tín hiệu trên chuỗi xung T2 (Hình C). Trên CT khung chậu là những nốt tiêu xương với viền tăng sinh ở xương chậu trái và xương cùng phải (Hình D
Như vậy, các yếu tố chính hướng tới ung thư nội mạc mạch máu bao gồm:
- Nữ giới trưởng thành đang điều trị liệu pháp Estrogen
- Hình ảnh đa nốt ngoại vi với sự co rút lớp vỏ.
- Di căn phổi canxi hóa.
- Dấu hiệu cây kẹo (‘Lollipop sign’)
U lympho ác tính nguyên phát tại gan (primary hepatic lymphoma)
Đây là nhóm ung thư cực kỳ hiếm gặp, chiếm dưới 1% các trường hợp u lympho ngoài hạch. Định nghĩa u lympho ác tính nguyên phát tại gan nếu không có bất kỳ bằng chứng nào của sự phát triển khối u tại lách, hạch bạch huyết hoặc bất kì vị trí di căn xa nào khác tại thời điểm chẩn đoán [32]. Tỷ lệ nam:nữ = 3:1, lứa tuổi thường gặp là 50 [33].
Tại thời điểm chẩn đoán, triệu chứng lâm sàng thường không đặc hiệu, có thể chỉ là khó chịu vùng bụng và sốt. Có mối liên quan chặt chẽ giữa bệnh lý này với tình trạng suy giảm miễn dịch, viêm gan B, C, hội chứng suy giảm miễn dịch mắc phải HIV và virus Epstein – Barr [34].
Khoảng 60% các trường hợp khối u xuất hiện đơn độc, là những khối ranh giới rõ, kích thước >4cm với viền vỏ ngấm thuốc mạnh sau tiêm, ít khi có tình trạng canxi hóa, gần tương tự dạng di căn tăng sinh mạch từ ống tiêu hóa, tuy nhiên những trường hợp này cần phát hiện thấy ung thư nguyên phát. Đa hạch và thâm nhiễm ít khi quan sát được và thường gắn với tình trạng di căn gan sau u Lympho hệ thống [34].
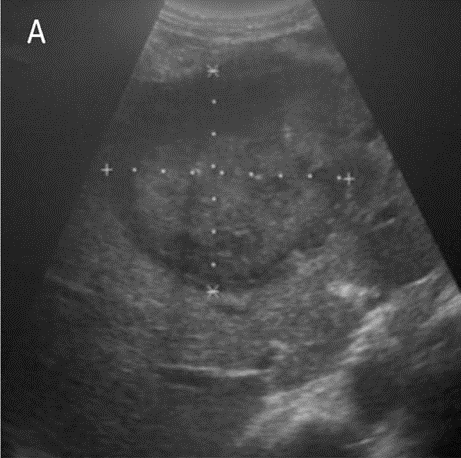

Hình 22: Primary Hepatic Lymphoma. Hình ảnh siêu âm cho thấy khối âm vang hỗn hợp tại vùng gianh giới giữa thùy gan trái và phải (Hình A). Kết hợp với hình ảnh CT là khối dạng vách xếp chồng lên nhau sau tiêm thuốc (Hình B).


Hình 23: MRI với hình ảnh khối u tín hiệu hỗn hợp thùy gan phải, với thương tổn mục tiêu và giảm tín hiệu trên chuỗi xung T1 (Hình A). Tăng tín hiệu tương đối trên chuỗi xung T2 (Hình B).
Tóm lại, một số yếu tố hướng tới chẩn đoán u lympho nguyên phát tại gan là:
- Không có tình trạng phát triển u lympho ngoài hạch ở các cơ quan khác.
- Khối u đơn độc với vỏ dày tăng ngấm thuốc
- Dấu hiệu cận lâm sàng điển hình nhưng không phát hiện vị trí ung thư nguyên phát.
- Bệnh nhân suy giảm miễn dịch
U thần kinh nội tiết nguyên phát tại gan (primary hepatic neuroendocrine tumor)
U thần kinh nội tiết nguyên phát tại gan (PHNT) chiếm dưới 0.3% trong tất cả các trường hợp u thần kinh nội tiết, chủ yếu xuất hiện ở nhóm bệnh nhân nữ 70 tuổi [32]. Triệu chứng lâm sàng không đặc hiệu, chủ yếu hay gặp đau bụng, mệt mỏi và gầy sút cân. Nhiều hơn 10% các trường hợp là không có triệu chứng lâm sàng, và chỉ có một tỷ lệ nhỏ bệnh nhân có hội chứng cận u (Cơn đỏ bừng mặt, đau đầu và đi ngoài) [35].
Chẩn đoán loại u này dựa trên giải phẫu bệnh và hóa mô miễn dịch, đồng thời loại trừ trường hợp u di căn. Chụp nhấp nháy đồ với Somatostatin là một kỹ thuật chẩn đoán cần thiết đối với u thần kinh nội tiết, với độ nhạy lên tới 90%. Mặt khác, PET/CT là phương thức chẩn đoán hình ảnh hay được sử dụng cho những khối u không có chức năng.
Hình ảnh trên cắt lớp vi tính cho thấy hình ảnh khối u đặc đơn độc hoặc đa u, các nốt tăng sinh mạch xen kẽ với các vị trí hoại tử hoặc canxi hóa.




Hình 24. Primary hepatic neuroendocrine carcinoma. CT không tiêm thuốc (Hình A) và sau tiêm thuốc (Hình B, C, D) cho thấy hình ảnh khối hỗn hợp, tăng sinh mạch tương đối với một vài nốt vôi hóa và các nang xen kẽ, nằm ở thùy gan trái.
Trên cộng hưởng từ là hình ảnh các khối u giảm tín hiệu trên chuỗi xung T1 và tăng tín hiệu trên chuỗi xung T2




Hình 25: Primary hepatic neuroendocrine carcinoma. MRI trên chuỗi xung T1 (Hình A), T2 (Hình B), và chuỗi xung tăng đối quang (Hình C) cho thấy khối tín hiệu hỗn hợp, giảm tín hiệu trên T1, tăng tín hiệu trên T2 xen kẽ các nang tín hiệu dịch. Khối u thể hiện tính chất kháng nước trên chuỗi xung Diffusion (Hình D
Chẩn đoán phân biệt thường rất đa dạng, một số khả năng có thể đặt ra là di căn gan loại tăng sinh mạch, ung thư biểu mô tế bào gan, ung thư đường mật trong gan với các triệu chứng thần kinh nội tiết, và dạng biểu mô của ung thư mô liên kết đường tiêu hóa (gastrointestinal stromal tumor – GIST) [35].
Phẫu thuật lấy khối u hoặc nhiều khối u, hóa chất động mạch gan (transcatheter arterial chemoembolization – TACE), là lựa chọn điều trị hỗ trợ tuyệt vời đối với những trường hợp tái phát [36].
Tóm lại, một số yếu tố chính hướng tới u thần kinh nội tiết nguyên phát tại gan là:
- Phụ nữ tuổi 60-70.
- Khối u tăng sinh mạch hoặc bình thường, không có tiền sử bệnh gan trước đó
- Có thể kết hợp với hội chứng cận u.
U tế bào nhỏ dạng tròn đa hình (desmoplastic small round cell tumor – DSCT)
U tế bào nhỏ dạng tròn đa hình (DSCT) là một khối u trung mô hiếm gặp và có độ xâm lấn mạnh, với khoảng 200 ca bệnh đã được mô tả trong y văn. Tỷ lệ gặp cao nhất ở nhóm vị thành niên (tuổi trung bình là 22), hay gặp ở nam giới (tỷ lệ nam : nữ là 4:1) [37]. Vị trí phát hiện nằm toàn bộ trong ổ bụng, không có một nguồn gốc tạng nhất định, xâm lấn vào mạc nối và phúc mạc bao gồm cả cơ hoành, cuống lách, dây chằng ruột cà phúc mạc đáy chậu [38]. Vị trí di căn hay gặp nhất là phổi và gan, bên cạnh phúc mạc.
Triệu chứng lâm sàng của DSCT ổ bụng không đặc hiệu, thường là các triệu chứng mơ hồ tại bụng hoặc khung chậu, đôi khi là sờ thấy khối u [38].
Trên chẩn đoán hình ảnh là hình ảnh khối u trong ổ bụng không rõ nguồn gốc tạng, phối hợp với nhiều nốt phúc mạc và dịch ổ bụng. Trên cắt lớp vi tính và cộng hưởng từ, khối u thể hiện hình ảnh khối tỷ trọng tổ chức, hoại tử trung tâm và chảy máu, ngấm thuốc vừa phải sau tiêm, canxi hóa trong 22% trường hợp [38].




Hình 26: Desmoplastic small round cell tumor. Hình ảnh CT thì tĩnh mạch cửa (Hình A, B) cho thấy hình ảnh giảm tín hiệu, giảm sinh mạch, xâm lấn cuống gan cùng với giãn đường mật trong gan trái thứ phát. Hình ảnh MRI chuỗi xung T1 và T2 xóa mỡ (Hình D) cho thấy khối giảm tín hiệu trên T1 và tăng tín hiệu trên T2 xâm lấn tới thành cuống gan
Tóm lại, trên một bệnh nhân trẻ tuổi, có một khối u trong ổ bụng với tình trạng xâm lấn phúc mạc rõ, không xác định được vị trí nguyên phát, thì có thể nghĩ tới DSCT.
KẾT LUẬN
Như vậy, có thể thấy bên cạnh ung thư biểu mô tế bào gan, về mặt giải phẫu bệnh, có rất nhiều loại ung thư ác tính khác nhau, có thể nguyên phát hoặc thứ phát. Mỗi loại ung thư có đặc điểm lâm sàng, cận lâm sàng khác nhau, đòi hỏi người thầy thuốc phải có sự tinh tế cũng như nhạy bén lâm sàng. Đồng thời, phải đẩy mạnh việc phối hợp với các chuyên ngành khác như chẩn đoán hình ảnh và đặc biệt là giải phẫu bệnh, đế chẩn đoán chính xác bệnh, với mục tiêu cuối cùng là điều trị thành công cho bệnh nhân.
TÀI LIỆU THAM KHẢO
1. Ros PR, Taylor HM (2000). Malignant tumors of the liver. In: Gore RM, Levine MS, eds. Textbook of gastrointestinal radiology, 2nd ed. Philadelphia: WB Saunders:1523–68
2. Jiang K., Al-Diffhala S., and Centeno B.A. (2018). Primary Liver Cancers-Part 1: Histopathology, Differential Diagnoses, and Risk Stratification. Cancer Control J Moffitt Cancer Cent, 25(1), 1073274817744625.
3. Schneider G., Grazioli L., and Saini S. (2006). Imaging of Malignant Focal Liver Lesions. MRI of the Liver: Imaging Techniques, Contrast Enhancement, Differential Diagnosis. Springer Milan, Milano, 187–235.
4. McFarland E.G., Mayo-Smith W.W., Saini S., et al. (1994). Hepatic hemangiomas and malignant tumors: improved differentiation with heavily T2-weighted conventional spin-echo MR imaging. Radiology, 193(1), 43–47.
5. Zhou Y.-M., Li B., Xu F., et al. (2008). Clinical features of hepatic angiomyolipoma. Hepatobiliary Pancreat Dis Int HBPD INT, 7(3), 284–287.
6. Du S., Li Y., Mao Y., et al. (2012). Diagnosis and treatment of hepatic angiomyolipoma. Hepatobiliary Surg Nutr, 1(1), 19–24.
7. Kobayashi Y., Kamimura K., Nomoto M., et al. (2013). Immunohistochemical Character of Hepatic Angiomyolipoma: For Its Management. Case Rep Med, 2013.
8. Mortelé K.J. and Ros P.R. (2001). Cystic focal liver lesions in the adult: differential CT and MR imaging features. Radiogr Rev Publ Radiol Soc N Am Inc, 21(4), 895–910.
9. Billington P.D., Prescott R.J., and Lapsia S. (2012). Diagnosis of a biliary cystadenoma demonstrating communication with the biliary system by MRI using a hepatocyte-specific contrast agent.Br J Radiol, 85(1010), e35-36.
10. Joel J.M., Jeyasingh S.D., and Kalyanaraman S. (2016). Biliary Cystadenoma: A Case Report. J Clin Diagn Res JCDR, 10(2), ED19-ED20.
11. Lewin M., Mourra N., Honigman I., et al. (2006). Assessment of MRI and MRCP in diagnosis of biliary cystadenoma and cystadenocarcinoma. Eur Radiol, 16(2), 407–413.
12. Lim J.H. (2004). Cholangiocarcinoma: recent advances in imaging and intervention. Abdom Imaging, 29(5), 538–539.
13. Fulcher A.S. and Sterling R.K. (2002). Hepatic neoplasms: computed tomography and magnetic resonance features. J Clin Gastroenterol, 34(4), 463–471.
14. Ohtsuka M., Ito H., Kimura F., et al. (2002). Results of surgical treatment for intrahepatic cholangiocarcinoma and clinicopathological factors influencing survival. Br J Surg, 89(12), 1525–1531.
15. Phạm Kim Bình (2015). Hình ảnh giải phẫu bệnh ung thư đường mật được mổ tại bệnh viện Việt-Đức từ 1-2001 đến 30-12-2004. Y Học Việt Nam, 310, 61–67.
16. Pedrassa B.C., da Rocha E.L., Kierszenbaum M.L., et al. (2014). Uncommon hepatic tumors: iconographic essay – Part 1. Radiol Bras, 47(5), 310–316.
17. Corrigan K. and Semelka R.C. (1995). Dynamic contrast-enhanced MR imaging of fibrolamellar hepatocellular carcinoma. Abdom Imaging, 20(2), 122–125.
18. Craig J.R., Peters R.L., Edmondson H.A., et al. (1980). Fibrolamellar carcinoma of the liver: a tumor of adolescents and young adults with distinctive clinico-pathologic features. Cancer, 46(2), 372–379.
19. Pinna A.D., Iwatsuki S., Lee R.G., et al. (1997). Treatment of Fibrolamellar Hepatoma With Subtotal Hepatectomy or Transplantation. Hepatol Baltim Md, 26(4), 877–883.
20. Que F.G. and Nagorney D.M. (1995). Hepatocellular Carcinoma: A Western Perspective. Dig Surg, 12(1), 45–52.
21. Ichikawa T., Federle M.P., Grazioli L., et al. (2000). Fibrolamellar hepatocellular carcinoma: pre- and posttherapy evaluation with CT and MR imaging. Radiology, 217(1), 145–151.
22. McLarney J.K., Rucker P.T., Bender G.N., et al. (1999). Fibrolamellar carcinoma of the liver: radiologic-pathologic correlation. Radiogr Rev Publ Radiol Soc N Am Inc, 19(2), 453–471.
23. Ichikawa T., Federle M.P., Grazioli L., et al. (1999). Fibrolamellar hepatocellular carcinoma: imaging and pathologic findings in 31 recent cases. Radiology, 213(2), 352–361.
24. Yu R.-S., Chen Y., Jiang B., et al. (2008). Primary hepatic sarcomas: CT findings. Eur Radiol, 18(10), 2196–2205.
25. Koyama T., Fletcher J.G., Johnson C.D., et al. (2002). Primary hepatic angiosarcoma: findings at CT and MR imaging. Radiology, 222(3), 667–673.
26. Kim H.R., Rha S.Y., Cheon S.H., et al. (2009). Clinical features and treatment outcomes of advanced stage primary hepatic angiosarcoma. Ann Oncol, 20(4), 780–787.
27. Giardino A., Miller F.H., Kalb B., et al. (2016). Hepatic epithelioid hemangioendothelioma: a report from three university centers. Radiol Bras, 49(5), 288–294.
28. Lyburn I.D., Torreggiani W.C., Harris A.C., et al. (2003). Hepatic epithelioid hemangioendothelioma: sonographic, CT, and MR imaging appearances. AJR Am J Roentgenol, 180(5), 1359–1364.
29. Choi K.H. and Moon W.S. (2013). Epithelioid hemangioendothelioma of the liver. Clin Mol Hepatol, 19(3), 315–319.
30. Kim K.A., Kim K.W., Park S.H., et al. (2006). Unusual mesenchymal liver tumors in adults: radiologic-pathologic correlation. AJR Am J Roentgenol, 187(5), W481-489.
31. Alomari A.I. (2006). The lollipop sign: a new cross-sectional sign of hepatic epithelioid hemangioendothelioma. Eur J Radiol, 59(3), 460–464.
32. Pedrassa B.C., da Rocha E.L., Kierzenbaum M.L., et al. (2014). Uncommon hepatic tumors: iconographic essay – Part 2. Radiol Bras, 47(6), 374–379.
33. Patel T.S., Malvania R., Shah M.C., et al. (2015). Primary hepatic lymphoma: A case report. J Cytol Indian Acad Cytol, 32(1), 36–38.
34. Tan Y. and Xiao E. (2013). Rare hepatic malignant tumors: dynamic CT, MRI, and clinicopathologic features: with analysis of 54 cases and review of the literature. Abdom Imaging, 38(3), 511–526.
35. Gao J., Hu Z., Wu J., et al. (2011). Primary hepatic carcinoid tumor. World J Surg Oncol, 9, 151.
36. Park C.H., Chung J.W., Jang S.J., et al. (2012). Clinical features and outcomes of primary hepatic neuroendocrine carcinomas. J Gastroenterol Hepatol, 27(8), 1306–1311.
37. Dufresne A., Cassier P., Couraud L., et al. (2012). Desmoplastic Small Round Cell Tumor: Current Management and Recent Findings. Sarcoma, 2012.
38. Pickhardt P.J., Fisher A.J., Balfe D.M., et al. (1999). Desmoplastic small round cell tumor of the abdomen: radiologic-histopathologic correlation. Radiology, 210(3), 633–638.
